For centuries, sickle cell disease operated on a brutal, microscopic logic. A single point mutation in the beta-globin gene—an adenine where there should be a thymine—caused hemoglobin molecules to polymerize under stress. They crystallized. Red blood cells, normally pliant discs, deformed into rigid crescents. These cells clogged capillaries, causing waves of ischemic pain known as crises. They died early, leading to profound anemia. The body was in a constant state of civil war.
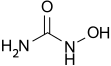
Hydroxyurea, known chemically as hydroxycarbamide, was not new. Synthesized in 1869, it found its first major use as a myelosuppressive agent in cancer chemotherapy. Its mechanism was straightforward: it inhibited ribonucleotide reductase, slowing DNA synthesis. In the late 1980s, researchers observed a secondary effect. In patients with sickle cell disease, the drug modestly increased the production of fetal hemoglobin—a form we normally stop making shortly after birth. Fetal hemoglobin lacks the beta subunit that carries the mutation. It acts as a molecular diluent, preventing the sickling polymerization.
The FDA’s approval on January 30, 1995, was not for a cure. It was for prevention. It sanctioned a chemical intervention that could reduce the frequency of excruciating pain crises by nearly half in many adults. The scale of the change was not measured in dramatic recoveries, but in quiet statistics: fewer hospitalizations, fewer days lost. It represented a shift from managing catastrophic failure to maintaining a fragile, biochemical equilibrium. It was the first pharmacological acknowledgment that the root of this ancient suffering could be gently, partially, persuaded to stand down.